About us
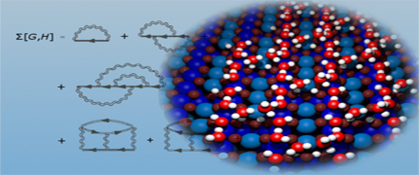
Computational materials science is one of the fastest developing fields in physics and chemistry. It focuses on the study of the complex properties of solids and liquids at the atomistic scale - making a quantum mechanical description of the interaction between atoms and electrons essential.
Although the basic concepts of quantum mechanics were discovered some 80 years ago, it's only in the last few decades that quantum mechanics has become widely used in materials science. Thanks to Walter Kohn's pioneering work on density functional theory (DFT), simulations of systems of several hundred atoms have become commonplace on modern computing platforms.
The Vienna ab-initio Simulations Package (VASP), developed in our group, is the most widely used DFT code for solids. VASP is currently used by more than 4000 research groups in industry and academia worldwide. It is also used as a common tool in most of the research projects in our group, allowing us to tackle systems of several thousand atoms on high performance parallel computers.
For further development, we focus on modern methods derived from quantum field theory and quantum chemistry, as well as novel machine learning techniques.
Using machine learning techniques, we can now extend modelling to systems of millions of atoms reaching the relevant time and length scales for real world applications.