Such calculations are computationally exceedingly demanding since in order to predict accurate melting temperatures, the free energy of the solid and the liquid must be calculated with very high precision, typically to a tolerance of about 1 meV per atom.
This requires stringent convergence tests as well as carefully laid out procedures in order to obtain sufficiently accurate absolute energies. A second, equally important issue is that the presently available density functionals are not accurate enough to yield very reliable predictions for the melting temperature. A prime example is the melting of silicon, which the local density approximation predicts to occur between 1300 and 1350 K, almost 20 % below the experimental melting point at 1687 K.
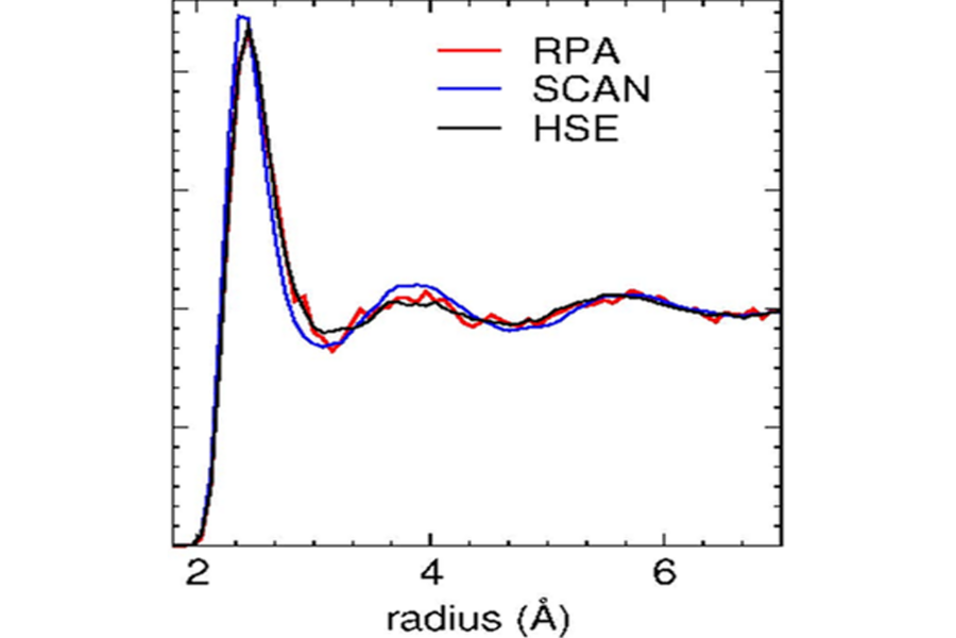
In this work the random phase approximation (RPA) an approximate many body technique, which includes exact exchange and the so called ring diagrams, is used to determine the silicon melting point. A melting temperature of about 1735 K and 1640 K without and with core polarization effects, respectively, is calculated bracketing the experimental melting temperature of 1687 K.
PHYSICAL REVIEW LETTERS 121, 195701, DOI: 10.1103/PhysRevLett.121.195701, P02 & P03