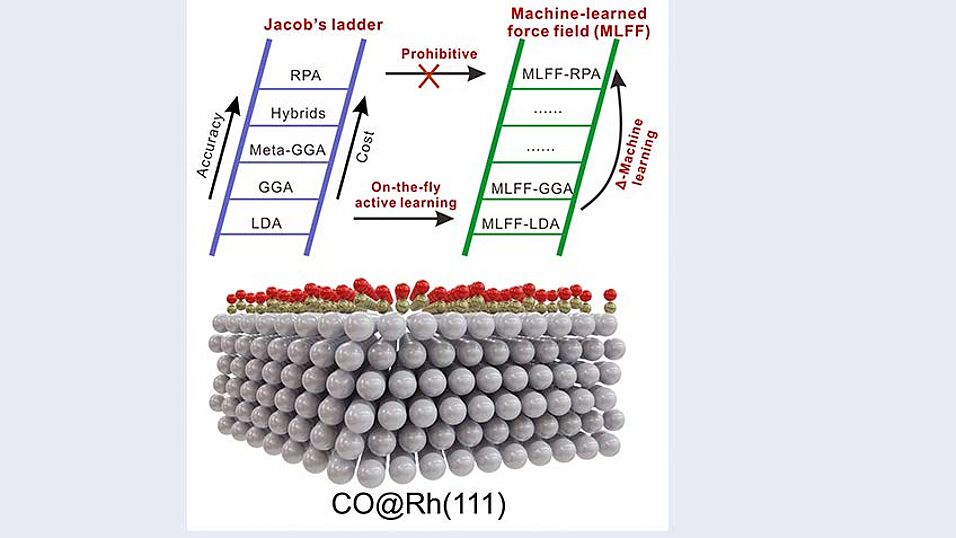
Machine learning allows us to explore the configuration space much faster and more thoroughly than before. This enables theoretical studies that we could only have dreamed of 10 years ago. What is holding us back now is that the theories we use to train machine learning models are based on density functional theory, which is itself an approximation. This can lead to incorrect predictions.
In this work, we correct for this error by learning the difference between a high-level many-body approach (the random phase approximation) and density functional theory. This correction allows us, for the first time, to accurately predict how carbon monoxide interacts with the Rh surface, finding excellent agreement with the experiment.
Our study paves the way for truly predictive “in silico” surface science and theoretical catalysis research. This research work was carried out jointly between the SFB TACO (University of Vienna) and the IMR, Chinese Academy of Sciences.
Abstract:
Adsorption of carbon monoxide (CO) on transition-metal surfaces is a prototypical process in surface sciences and catalysis. Despite its simplicity, it has posed great challenges to theoretical modeling. Pretty much all existing density functionals fail to accurately describe surface energies and CO adsorption site preference as well as adsorption energies simultaneously. Although the random phase approximation (RPA) cures these density functional theory failures, its large computational cost makes it prohibitive to study the CO adsorption for any but the simplest ordered cases. Here, we address these challenges by developing a machine-learned force field (MLFF) with near RPA accuracy for the prediction of coverage-dependent adsorption of CO on the Rh(111) surface through an efficient on-the-fly active learning procedure and a Δ-machine learning approach. We show that the RPA-derived MLFF is capable to accurately predict the Rh(111) surface energy and CO adsorption site preference as well as adsorption energies at different coverages that are all in good agreement with experiments. Moreover, the coverage-dependent ground-state adsorption patterns and adsorption saturation coverage are identified.
“Combining Machine Learning and Many-Body Calculations: Coverage-Dependent
Adsorption of CO on Rh(111)”,
Peitao Liu, Jiantao Wang, Noah Avargues, Carla Verdi, Andreas Singraber, Fer-
enc Karsai, Xing-Qiu Chen, and Georg Kresse Phys. Rev. Lett. 130, 078001
(2023).
The full article can be found here:
DOI: 10.1103/PhysRevLett.130.078001