The authors Stefan Riemelmoser, Carla Verdi, Merzuk Kaltak and Georg Kresse, have used machine learning to speed up electronic structure calculations. Their method has been implemented in the VASP code, and the training data is freely available at https://doi.org/10.25365/phaidra.418 .
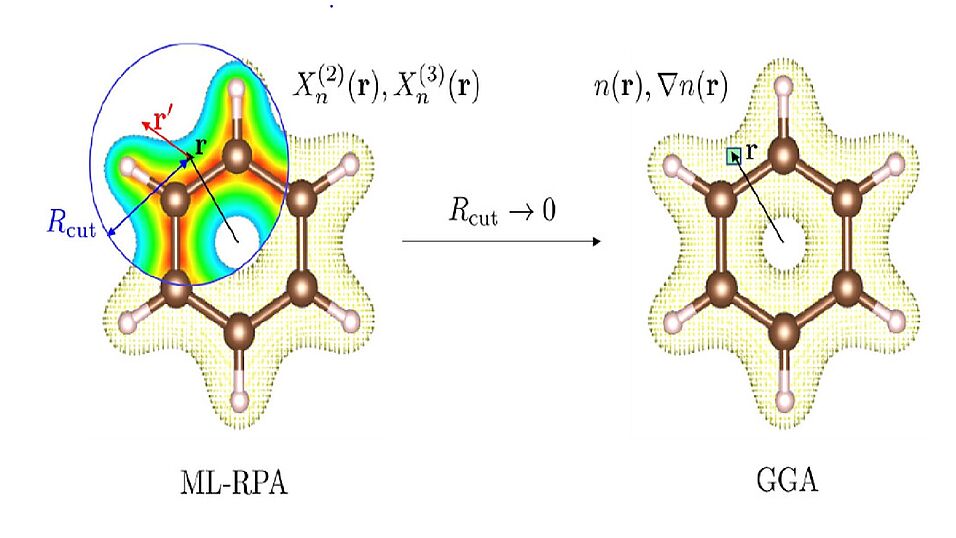
Kohn-Sham density functional theory (DFT) is the standard method for first-principles calculations in computational chemistry and materials science. More accurate theories such as the random-phase approximation (RPA) are limited in application due to their large computational cost. Here, we use machine learning to map the RPA to a pure Kohn–Sham density functional. The machine learned RPA model (ML-RPA) is a nonlocal extension of the standard gradient approximation. The density descriptors used as ingredients for the enhancement factor are nonlocal counterparts of the local density and its gradient. Rather than fitting only RPA exchange-correlation energies, we also include derivative information in the form of RPA optimized effective potentials. We train a single ML-RPA functional for diamond, its surfaces, and liquid water. The accuracy of ML-RPA for the formation energies of 28 diamond surfaces reaches that of state-of-the-art van der Waals functionals. For liquid water, however, ML-RPA cannot yet improve upon the standard gradient approximation. Overall, our work demonstrates how machine learning can extend the applicability of the RPA to larger system sizes, time scales, and chemical spaces.